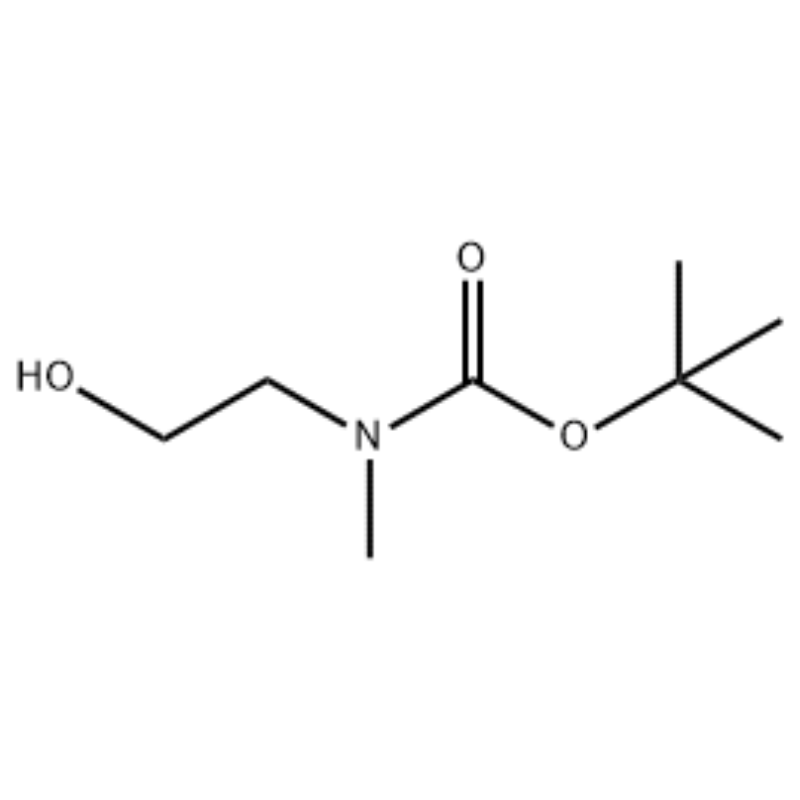
Til en opløsning af 2-(methylamino)ethanol (500 mg, 0,53 ml, 6,66 mmol) i CH2Cl2 (20 ml) blev der tilsat Boc2O (1,48 g, 6,79 mmol), efterfulgt af omrøring ved stuetemperatur i 1 time.Reaktionsopløsningen blev ekstraheret med saltvand og CH2Cl2.Det således opnåede organiske lag blev tørret over MgS04 og filtreret.Derefter blev filtratet koncentreret i vakuum til opnåelse af den omhandlede forbindelse (farveløs olie, kvantitativ);1H NMR (200 MHz, CDCI3) delta 3,74 (q, J= 10,5, 5,2 Hz, 2H) 3,25 (t, J= 5,2 Hz, 2H) 2,91 (s, 3H) 1,45 (s, 9H);massespektrum m/e (relativ intensitet) 144 (20) 102 (24) 57 (70) 44 (100).
Eksempel 38;N1-(3-fluor-4-(2-(1-(2-(methylamino)ethyl)-1H-imidazol-4-yl)thieno[3,2-b]pyridin-7-yloxy)phenyl)-N3 -(2-methoxyphenyl)malonamid (96);Trin 1: tert-butyl-2-hydroxyethyl(methyl)carbamat (97) (J. Med. Chem., 1999, 42, 11, 2008) Til en opløsning af 2-(methylamino)ethanol (5,0 g, 67 mmol) i THF (50 ml) ved stuetemperatur blev tilsat Boc2O (15,7 g, 72 mmol), og reaktionsblandingen blev omrørt ved stuetemperatur i 4 timer.Reaktionsblandingen blev koncentreret til tørhed, og titelforbindelsen 97 blev anvendt direkte i det næste trin uden yderligere oprensning (11,74 g, 100 % udbytte).MS (m/z): 176,2 (M+H).
Fremstilling af l-2-[4-brom-2-(4-oxo-2-ftiotaioxo1hiazolidin-5-ylidenmefliyl)phenoxy]efliyl-3-efliyl-l-methylurinstof(Compoiotamd 161) Trin 1: Syntese af t-butyl2- hydroxyethylmethylcarbamat;Til en opløsning af 2-(methylamino)ethanol (500 mg, 0,53 ml, 6,66 mmol) i CH2Cl2 (20 ml) blev der tilsat BoC2O (1,48 g, 6,79 mmol), efterfulgt af omrøring ved stuetemperatur i 1 time.Reaktionsopløsningen blev ekstraheret med saltvand og CH2Cl2.Det således opnåede organiske lag blev tørret over MgS04 og filtreret.Derefter blev filtratet koncentreret i vakuum for at opnå den omhandlede forbindelse (farveløs olie, kvantitativ); 1HNMR (200 MHz, CDCI3) delta 3,74 (q, J= 10,5, 5,2 Hz, 2H) 3,25 (t, J= 5,2 Hz, 2H) 2,91 (s, 3H) 1,45 (s, 9H);massespektrum m/e (relativ intensitet) 144 (20) 102 (24) 57 (70) 44 (100).
2-(methylamino)ethanol (90,1 g, 1,2 mol) blev opløst i 1,2 L methylenchlorid, og BoC2O (218 g, 1 mol) blev langsomt tilsat dertil under omrøring ved 0°C, efterfulgt af ved stuetemperatur i 3 timer.Reaktionsblandingen blev sekventielt vasket med 700 ml af en vandig opløsning af mættet ammoniumchlorid og 300 ml vand.Den vaskede blanding blev dehydreret under anvendelse af vandfrit natriumsulfat og koncentreret under reduceret tryk for at opnå forbindelsen (a) (175 g, 1 mol, 100%) som en olie uden farve. TLC: Rf = 0,5 (50% EtOAc i Hex) visualiseret med Ce-Mo farve1H NMR (600MHz, CDCl3) delta 1,47 (s, 9H), 2,88 (br s, IH), 3,41 (br s, 2H), 3,76 (br s, 2H).
90,1 g (1,2 mol) 2-(methylamino)ethanol blev opløst i 1,2 L methylenchlorid, 218 g (1 mol) Boc2O blev langsomt tilsat dertil, mens den resulterende opløsning blev omrørt ved 0C, og den resulterende opløsning blev omrørt ved stuetemperatur i 3 timer.Reaktionsblandingen blev sekventielt vasket med 700 ml af en vandig mættet ammoniumchloridopløsning og 300 ml vand, dehydreret under anvendelse af vandfrit natriumsulfat og derefter koncentreret under reduceret tryk for at opnå 175 g (1 mol) af en akromatisk olieforbindelse beskyttet af Boc-gruppe (udbytte: 100%).1H NMR (600 MHz, CDCI3) delta 7,84 (br s, 2H), 7,76 (br s, 2H), 4,34 (d, J = 15,0 Hz, 2H), 3,63 (br s, 2H), 3,04 (d , J = 15,0 Hz, 3H), 1,46 (d, J = 16,2 Hz, 9H) 90 g (0,514 mol) af den opnåede forbindelse blev opløst i 1,5 L tetrahydrofuran, 88,0 g (539 mol) N- hydroxyphthalimid og 141 g (0,539 mol) triphenylphosphin blev tilsat dertil, 106 ml (0,539 mol) diisopropylazodicarboxylat blev langsomt tilsat dertil under omrøring af den resulterende opløsning ved 0°C, og den resulterende opløsning blev omrørt i 3 timer, mens temperaturen deraf blev hævet. til stuetemperatur.Efter koncentrering af reaktionsblandingen under reduceret tryk blev 600 ml isopropylether tilsat dertil, den resulterende opløsning blev omrørt ved 0°C i 1 time, og hvid fast type triphenylphosphinoxid blev filtreret.Det faste stof blev vasket med 200 ml isopropylether afkølet til 0°C og opsamlet med det første filtrat, og det resulterende filtrat blev koncentreret under reduceret tryk til opnåelse af 198 g af en blanding af forbindelse XX og diisopropylhydrazodicarboxylat i et blandingsforhold på 10 til 15 %. (udbytte: 120%).1H NMR (600 MHz, CDCl3) delta 7,84 (br s, 2H), 7,76 (br s, 2H), 4,34 (d, J = 15,0 Hz, 2H), 3,63 (br s, 2H), 3,04 (d J = 15,0 Hz, 3H), 1,46 (d, J= 16,2 Hz, 9H)
















 Bygning 12, nr. 309, South 2nd Road, Economic Development Zone, Longquanyi District, Chengdu, Sichuan, Kina.
Bygning 12, nr. 309, South 2nd Road, Economic Development Zone, Longquanyi District, Chengdu, Sichuan, Kina. amy@enlaibio.com / cynthia@enlaibio.com / edison@enlaibio.com / daisy@enlaibio.com
amy@enlaibio.com / cynthia@enlaibio.com / edison@enlaibio.com / daisy@enlaibio.com +86 (028) 84841969
+86 (028) 84841969 +86 135 5885 5404
+86 135 5885 5404



.png)


